
MDM2 Inhibition in the Treatment of Glioblastoma: From Concept to Clinical Investigation

Abstract
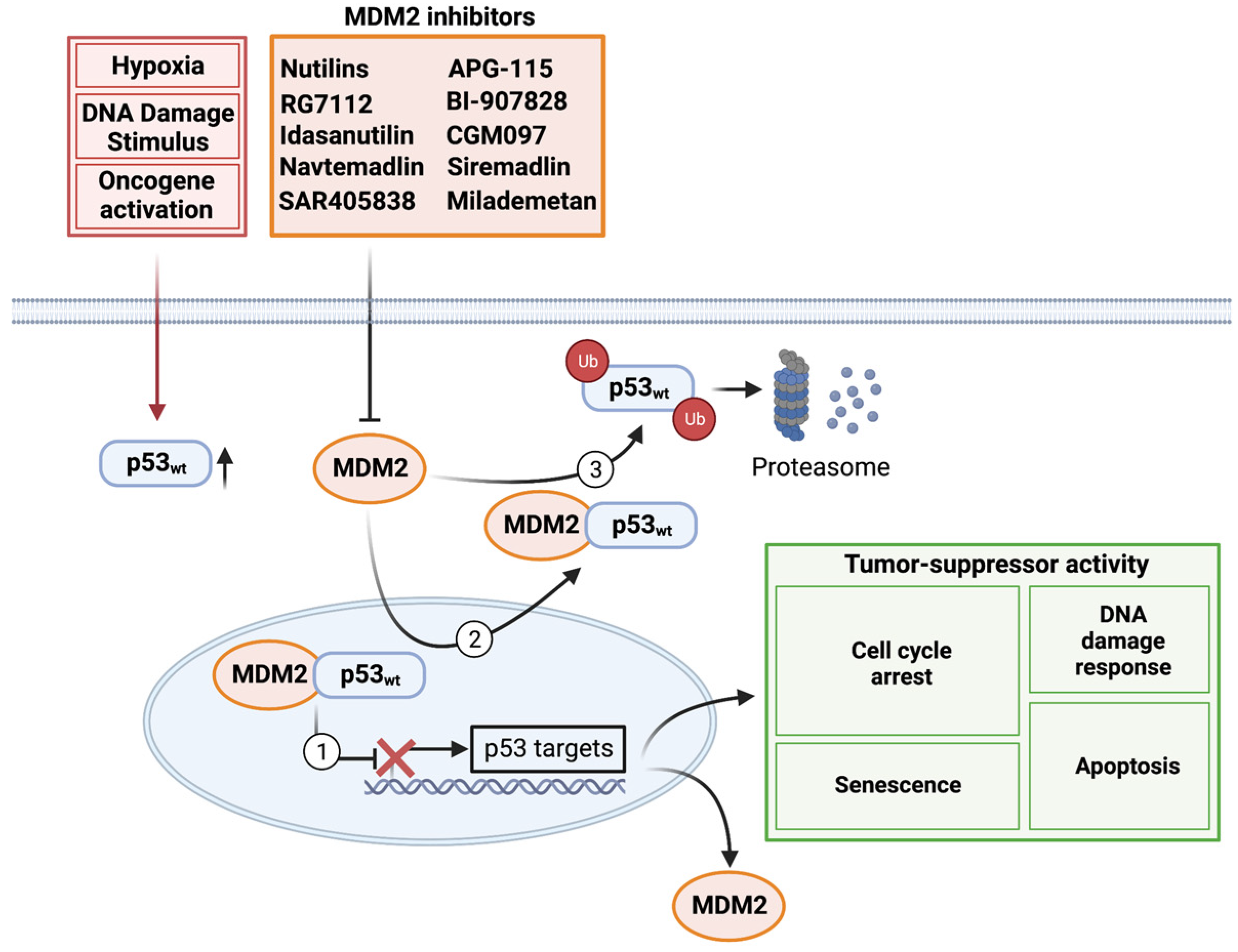
:1. Introduction
2. Basic Biology
3. Preclinical Evaluation and Clinical Development of MDM2 Inhibitors
3.1. Nutilins
3.2. RG7112 (RO5045337) and Idasanutlin (RG7388)
3.3. Navtemadlin (KRT-232/AMG-232)
3.4. SAR405838 and APG-115
3.5. BI-907828
3.6. CGM097
3.7. Siremadlin (HDM201)
3.8. Milademetan (DS-3032b)
3.9. ALRN-6924 (Dual MDM2/MDMX Inhibitor)
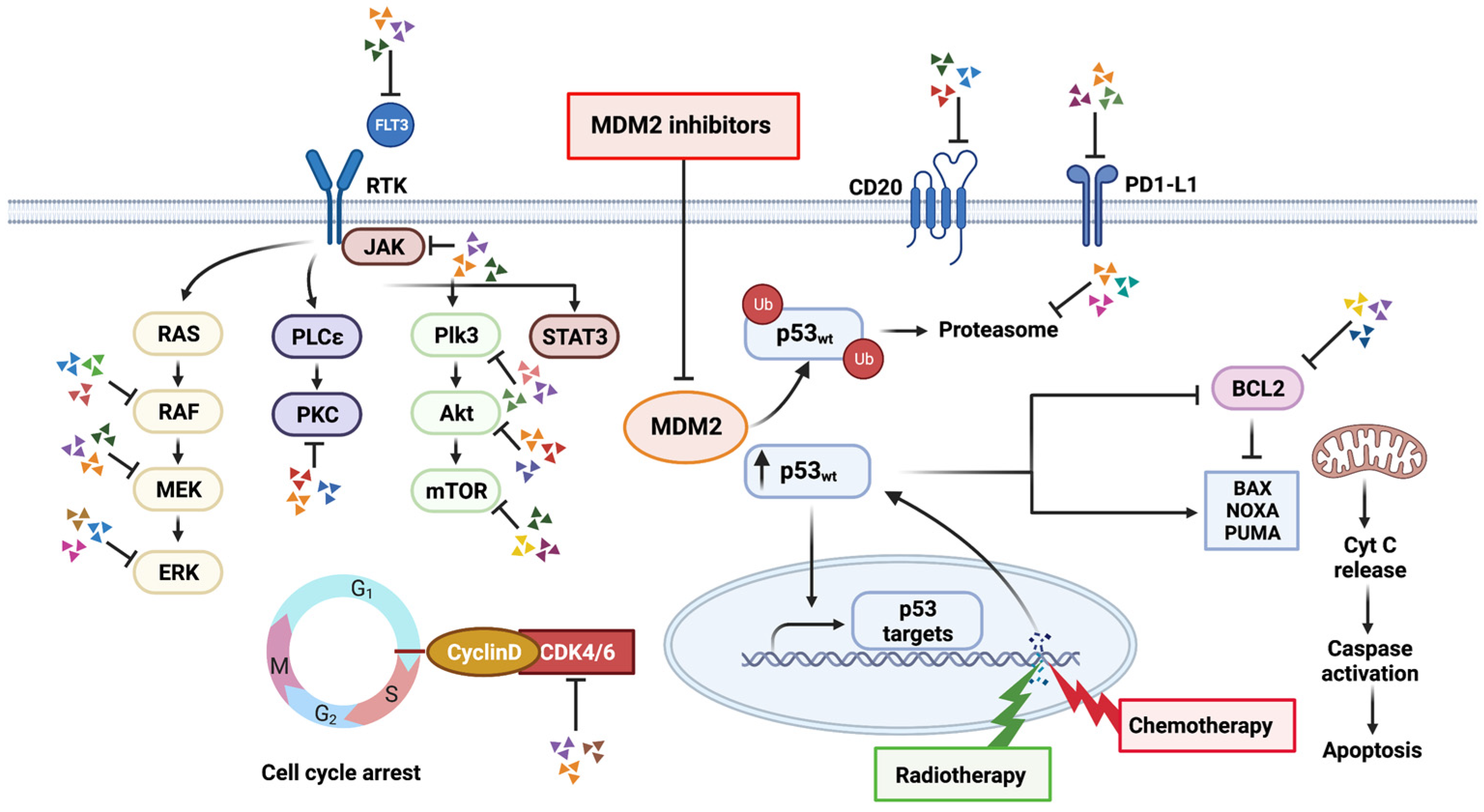
4. Combination Therapy
4.1. Radiation Therapy
4.2. Chemotherapy
4.3. Potential Side Effects and Toxicity Profile
4.4. Targeted Therapy
5. Clinical Trials
5.1. NCT01723020
5.2. NCT03107780
5.3. NCT03158389
5.4. NCT03654716
5.5. NCT05376800
5.6. Phase 3 Trials in Other Cancers
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Carrano, A.; Juarez, J.J.; Incontri, D.; Ibarra, A.; Guerrero Cazares, H. Sex-Specific Differences in Glioblastoma. Cells 2021, 10, 1783. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cote, D.J.; Ascha, M.; Kruchko, C.; Barnholtz-Sloan, J.S. Adult Glioma Incidence and Survival by Race or Ethnicity in the United States From 2000 to 2014. JAMA Oncol. 2018, 4, 1254–1262. [Google Scholar] [CrossRef][Green Version]
- Fyllingen, E.H.; Bø, L.E.; Reinertsen, I.; Jakola, A.S.; Sagberg, L.M.; Berntsen, E.M.; Salvesen, Ø.; Solheim, O. Survival of glioblastoma in relation to tumor location: A statistical tumor atlas of a population-based cohort. Acta Neurochir. 2021, 163, 1895–1905. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Price, M.; Ryan, K.; Edelson, J.; Neff, C.; Cioffi, G.; Waite, K.A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Pediatric Brain Tumor Foundation Childhood and Adolescent Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2022, 24, iii1–iii38. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef][Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef][Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef][Green Version]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Collins, V.P. Mechanisms of disease: Genetic predictors of response to treatment in brain tumors. Nat. Clin. Pract. Oncol. 2007, 4, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nature reviews. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Feldser, D.M.; Kostova, K.K.; Winslow, M.M.; Taylor, S.E.; Cashman, C.; Whittaker, C.A.; Sanchez-Rivera, F.J.; Resnick, R.; Bronson, R.; Hemann, M.T.; et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature 2010, 468, 572–575. [Google Scholar] [CrossRef][Green Version]
- Dickins, R.A.; Hemann, M.T.; Zilfou, J.T.; Simpson, D.R.; Ibarra, I.; Hannon, G.J.; Lowe, S.W. Probing Tumor Phenotypes Using Stable and Regulated Synthetic microRNA Precursors. Nat. Genet. 2006, 38, 389. [Google Scholar] [CrossRef]
- Stewart-Ornstein, J.; Lahav, G. p53 dynamics in response to DNA damage vary across cell lines and are shaped by efficiency of DNA repair and activity of the kinase ATM. Sci. Signal. 2017, 10, eaah6671. [Google Scholar] [CrossRef][Green Version]
- Jiang, L.; Sheikh, M.S.; Huang, Y. Decision Making by p53: Life versus Death. Mol. Cell. Pharmacol. 2010, 2, 69–77. [Google Scholar]
- Wu, L.; Zhou, N.; Sun, R.; Chen, X.D.; Feng, S.C.; Zhang, B.; Bao, J.K. Network-based identification of key proteins involved in apoptosis and cell cycle regulation. Cell Prolif. 2014, 47, 356–368. [Google Scholar] [CrossRef]
- Wu, M.; Ye, H.; Tang, Z.; Shao, C.; Lu, G.; Chen, B.; Yang, Y.; Wang, G.; Hao, H. p53 dynamics orchestrates with binding affinity to target genes for cell fate decision. Cell Death Dis. 2017, 8, e3130. [Google Scholar] [CrossRef][Green Version]
- Wang, P.; Guan, D.; Zhang, X.P.; Liu, F.; Wang, W. Modeling the regulation of p53 activation by HIF-1 upon hypoxia. FEBS Lett. 2019, 593, 2596–2611. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. The p53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu. Rev. Biochem. 2016, 85, 375–404. [Google Scholar] [CrossRef]
- Vassilev, L.T. MDM2 inhibitors for cancer therapy. Trends Mol. Med. 2007, 13, 23–31. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nature reviews. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef][Green Version]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef][Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e411. [Google Scholar] [CrossRef][Green Version]
- Guo, C.F.; Zhuang, Y.; Chen, Y.; Chen, S.; Peng, H.; Zhou, S. Significance of tumor protein p53 mutation in cellular process and drug selection in brain lower grade (WHO grades II and III) glioma. Biomark Med. 2020, 14, 1139–1150. [Google Scholar] [CrossRef]
- Noor, H.; Briggs, N.E.; McDonald, K.L.; Holst, J.; Vittorio, O. TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy. Cancers 2021, 13, 5362. [Google Scholar] [CrossRef] [PubMed]
- Marker, D.F.; Agnihotri, S.; Amankulor, N.; Murdoch, G.H.; Pearce, T.M. The dominant TP53 hotspot mutation in IDH -mutant astrocytoma, R273C, has distinctive pathologic features and sex-specific prognostic implications. Neurooncol. Adv. 2022, 4, vdab182. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef][Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Xu, Z.; Scuoppo, C.; Rillahan, C.D.; Gao, J.; Spitzer, B.; Bosbach, B.; Kastenhuber, E.R.; Baslan, T.; et al. Deletions linked to TP53 loss drive cancer through p53-independent mechanisms. Nature 2016, 531, 471–475. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kato, S.; Han, S.Y.; Liu, W.; Otsuka, K.; Shibata, H.; Kanamaru, R.; Ishioka, C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 8424–8429. [Google Scholar] [CrossRef]
- Devine, T.; Dai, M.-S. Targeting the ubiquitin-mediated proteasome degradation of p53 for cancer therapy. Curr. Pharm. Des. 2013, 19, 3248–3262. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Carter, S.; Bischof, O.; Dejean, A.; Vousden, K.H. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat. Cell Biol. 2007, 9, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol. 2016, 11, 617–644. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Hu, C.; Qutub, A.; Qiu, Y.H.; Zhang, X.; Post, S.M.; Zhang, N.; Coombes, K.; Kornblau, S.M. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia 2017, 31, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef][Green Version]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef][Green Version]
- Kato, S.; Ross, J.S.; Gay, L.; Dayyani, F.; Roszik, J.; Subbiah, V.; Kurzrock, R. Analysis of MDM2 Amplification: Next-Generation Sequencing of Patients with Diverse Malignancies. JCO Precis Oncol. 2018, 2018, PO.17.00235. [Google Scholar] [CrossRef]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2658 human cancer genomes. Cell 2021, 184, 2239–2254.e39. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Martinelli, G.; Daver, N.; Papayannidis, C.; Wei, A.; Higgins, B.; Ott, M.; Mascarenhas, J.; Andreeff, M. MDM2 inhibition: An important step forward in cancer therapy. Leukemia 2020, 34, 2858–2874. [Google Scholar] [CrossRef]
- Zhang, B.; Golding, B.T.; Hardcastle, I.R. Small-molecule MDM2-p53 inhibitors: Recent advances. Future Med. Chem. 2015, 7, 631–645. [Google Scholar] [CrossRef][Green Version]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef][Green Version]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.Y. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef][Green Version]
- Lakoma, A.; Barbieri, E.; Agarwal, S.; Jackson, J.; Chen, Z.; Kim, Y.; McVay, M.; Shohet, J.M.; Kim, E.S. The MDM2 small-molecule inhibitor RG7388 leads to potent tumor inhibition in p53 wild-type neuroblastoma. Cell Death Discov. 2015, 1, 15026. [Google Scholar] [CrossRef][Green Version]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussiere, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef][Green Version]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical Overview of MDM2/X-Targeted Therapies. Front. Oncol. 2016, 6, 7. [Google Scholar] [CrossRef][Green Version]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 133. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2022, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tovar, C.; Rosinski, J.; Filipovic, Z.; Higgins, B.; Kolinsky, K.; Hilton, H.; Zhao, X.; Vu, B.T.; Qing, W.; Packman, K.; et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 1888–1893. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef]
- Kojima, K.; Konopleva, M.; Samudio, I.J.; Shikami, M.; Cabreira-Hansen, M.; McQueen, T.; Ruvolo, V.; Tsao, T.; Zeng, Z.; Vassilev, L.T.; et al. MDM2 antagonists induce p53-dependent apoptosis in AML: Implications for leukemia therapy. Blood 2005, 106, 3150–3159. [Google Scholar] [CrossRef][Green Version]
- Villalonga-Planells, R.; Coll-Mulet, L.; Martínez-Soler, F.; Castaño, E.; Acebes, J.J.; Giménez-Bonafé, P.; Gil, J.; Tortosa, A. Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS ONE 2011, 6, e18588. [Google Scholar] [CrossRef][Green Version]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, M.; Wang, X.; Li, Y.; Tripodi, J.; Mosoyan, G.; Mascarenhas, J.; Kremyanskaya, M.; Najfeld, V.; Hoffman, R. Combination treatment in vitro with Nutlin, a small-molecule antagonist of MDM2, and pegylated interferon-α 2a specifically targets JAK2V617F-positive polycythemia vera cells. Blood 2012, 120, 3098–3105. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xia, L.; Li, Y.; Wang, X.; Hoffman, R. The orally bioavailable MDM2 antagonist RG7112 and pegylated interferon α 2a target JAK2V617F-positive progenitor and stem cells. Blood 2014, 124, 771–779. [Google Scholar] [CrossRef][Green Version]
- Ray-Coquard, I.; Blay, J.Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef][Green Version]
- Higgins, B.; Tovar, C.; Glenn, K.; Walz, A.; Filipovic, Z.; Zhang, Y.-E.; Dangl, M.; Graves, B.; Vassilev, L.; Packman, K. Abstract B55: Antitumor activity of the MDM2 antagonist RG7388. Mol. Cancer Ther. 2013, 12, B55. [Google Scholar] [CrossRef]
- Berberich, A.; Kessler, T.; Thomé, C.M.; Pusch, S.; Hielscher, T.; Sahm, F.; Oezen, I.; Schmitt, L.M.; Ciprut, S.; Hucke, N.; et al. Targeting Resistance against the MDM2 Inhibitor RG7388 in Glioblastoma Cells by the MEK Inhibitor Trametinib. Clin. Cancer Res. 2019, 25, 253–265. [Google Scholar] [CrossRef][Green Version]
- Siu, L.L.; Italiano, A.; Miller, W.H.; Blay, J.-Y.; Gietema, J.A.; Bang, Y.-J.; Mileshkin, L.R.; Hirte, H.W.; Reckner, M.; Higgins, B. Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist, in patients (pts) with solid tumors. J. Clin. Oncol. 2014, 32, 2535. [Google Scholar] [CrossRef]
- Higgins, B.; Glenn, K.; Walz, A.; Tovar, C.; Filipovic, Z.; Hussain, S.; Lee, E.; Kolinsky, K.; Tannu, S.; Adames, V.; et al. Preclinical optimization of MDM2 antagonist scheduling for cancer treatment by using a model-based approach. Clin. Cancer Res. 2014, 20, 3742–3752. [Google Scholar] [CrossRef][Green Version]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef]
- Canon, J.; Osgood, T.; Olson, S.H.; Saiki, A.Y.; Robertson, R.; Yu, D.; Eksterowicz, J.; Ye, Q.; Jin, L.; Chen, A.; et al. The MDM2 Inhibitor AMG 232 Demonstrates Robust Antitumor Efficacy and Potentiates the Activity of p53-Inducing Cytotoxic Agents. Mol. Cancer Ther. 2015, 14, 649–658. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Her, N.G.; Oh, J.W.; Oh, Y.J.; Han, S.; Cho, H.J.; Lee, Y.; Ryu, G.H.; Nam, D.H. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 2018, 9, 792. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef][Green Version]
- Kim, M.; Ma, D.J.; Calligaris, D.; Zhang, S.; Feathers, R.W.; Vaubel, R.A.; Meaux, I.; Mladek, A.C.; Parrish, K.E.; Jin, F.; et al. Efficacy of the MDM2 Inhibitor SAR405838 in Glioblastoma Is Limited by Poor Distribution Across the Blood-Brain Barrier. Mol. Cancer Ther. 2018, 17, 1893–1901. [Google Scholar] [CrossRef][Green Version]
- de Jonge, M.; de Weger, V.A.; Dickson, M.A.; Langenberg, M.; Le Cesne, A.; Wagner, A.J.; Hsu, K.; Zheng, W.; Macé, S.; Tuffal, G.; et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur. J. Cancer 2017, 76, 144–151. [Google Scholar] [CrossRef]
- de Weger, V.A.; de Jonge, M.; Langenberg, M.H.G.; Schellens, J.H.M.; Lolkema, M.; Varga, A.; Demers, B.; Thomas, K.; Hsu, K.; Tuffal, G.; et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 286–293. [Google Scholar] [CrossRef][Green Version]
- Aguilar, A.; Lu, J.; Liu, L.; Du, D.; Bernard, D.; McEachern, D.; Przybranowski, S.; Li, X.; Luo, R.; Wen, B. Discovery of 4-((3′ R, 4′ S, 5′ R)-6 ″-Chloro-4′-(3-chloro-2-fluorophenyl)-1′-ethyl-2 ″-oxodispiro [cyclohexane-1, 2′-pyrrolidine-3′, 3 ″-indoline]-5′-carboxamido) bicyclo [2.2.2] octane-1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development. J. Med. Chem. 2017, 60, 2819–2839. [Google Scholar] [PubMed]
- Skalniak, L.; Surmiak, E.; Holak, T.A. A therapeutic patent overview of MDM2/X-targeted therapies (2014-2018). Expert Opin. Ther. Pat. 2019, 29, 151–170. [Google Scholar] [CrossRef][Green Version]
- Chong, C.R.; Bauer, T.M.; Laurie, S.A.; Patel, M.R.; Yamamoto, N.; Davenport, T.; Geng, J.; Gibson, N.; Vallaster, M.P.; LoRusso, P. A phase Ia/Ib, open label, multicenter, dose-escalation study of BI 907828 (MDM2-p53 antagonist) in adult patients with advanced or metastatic solid tumors. J. Clin. Oncol. 2019, 37, TPS3166. [Google Scholar] [CrossRef]
- Hao, X.; Bahia, R.; Cseh, O.; Bozek, D.; Blake, S.; Rinnenthal, J.; Weyer-Czernilofsky, U.; Rudolph, D.; Luchman, H.A. BI-907828, a novel potent MDM2 inhibitor, inhibits GBM brain tumor stem cells in vitro and prolongs survival in orthotopic xenograft mouse models. Neuro-Oncol. 2023, 25, 913–926. [Google Scholar] [CrossRef]
- Cornillie, J.; Wozniak, A.; Li, H.; Gebreyohannes, Y.K.; Wellens, J.; Hompes, D.; Debiec-Rychter, M.; Sciot, R.; Schoffski, P. Anti-tumor activity of the MDM2-TP53 inhibitor BI-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring MDM2 amplification. Clin. Transl. Oncol. 2020, 22, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Rinnenthal, J.; Rudolph, D.; Blake, S.; Gollner, A.; Wernitznig, A.; Weyer-Czernilofsky, U.; Haslinger, C.; Garin-Chesa, P.; Moll, J.; Kraut, N. BI 907828: A highly potent MDM2 inhibitor with low human dose estimation, designed for high-dose intermittent schedules in the clinic. Cancer Res. 2018, 78, 4865. [Google Scholar] [CrossRef]
- Gessier, F.; Kallen, J.; Jacoby, E.; Chène, P.; Stachyra-Valat, T.; Ruetz, S.; Jeay, S.; Holzer, P.; Masuya, K.; Furet, P. Discovery of dihydroisoquinolinone derivatives as novel inhibitors of the p53-MDM2 interaction with a distinct binding mode. Bioorg. Med. Chem. Lett. 2015, 25, 3621–3625. [Google Scholar] [CrossRef]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Chen, X.Y.; Dong, X.D.; Wang, J.Q.; Feng, W.; Teng, Q.X.; Cui, Q.; Li, J.; Li, X.Q.; Chen, Z.S. NVP-CGM097, an HDM2 Inhibitor, Antagonizes ATP-Binding Cassette Subfamily B Member 1-Mediated Drug Resistance. Front. Oncol. 2020, 10, 1219. [Google Scholar] [CrossRef]
- Maser, T.; Zagorski, J.; Kelly, S.; Ostrander, A.; Goodyke, A.; Nagulapally, A.; Bond, J.; Park, Y.; Saulnier Sholler, G. The MDM2 inhibitor CGM097 combined with the BET inhibitor OTX015 induces cell death and inhibits tumor growth in models of neuroblastoma. Cancer Med. 2020, 9, 8144–8158. [Google Scholar] [CrossRef]
- Reuther, C.; Heinzle, V.; Nölting, S.; Herterich, S.; Hahner, S.; Halilovic, E.; Jeay, S.; Wuerthner, J.U.; Aristizabal Prada, E.T.; Spöttl, G.; et al. The HDM2 (MDM2) Inhibitor NVP-CGM097 Inhibits Tumor Cell Proliferation and Shows Additive Effects with 5-Fluorouracil on the p53-p21-Rb-E2F1 Cascade in the p53wild type Neuroendocrine Tumor Cell Line GOT1. Neuroendocrinology 2018, 106, 1–19. [Google Scholar] [CrossRef]
- Townsend, E.C.; DeSouza, T.; Murakami, M.A.; Montero, J.; Stevenson, K.; Christie, A.L.; Christodolou, A.N.; Vojinovic, U.; Kopp, N.; Barzaghi-Rinaudo, P. The MDM2 inhibitor NVP-CGM097 is highly active in a randomized preclinical trial of B-cell acute lymphoblastic leukemia patient derived xenografts. Blood 2015, 126, 797. [Google Scholar] [CrossRef]
- Holzer, P. Discovery of Potent and Selective p53-MDM2 Protein-Protein Interaction Inhibitors as Anticancer Drugs. Chimia 2017, 71, 716–721. [Google Scholar] [CrossRef]
- Ferretti, S.; Rebmann, R.; Berger, M.; Santacroce, F.; Albrecht, G.; Pollehn, K.; Sterker, D.; Wartmann, M.; Hueber, A.; Wiesmann, M. Insights into the mechanism of action of NVP-HDM201, a differentiated and versatile Next-Generation small-molecule inhibitor of Mdm2, under evaluation in phase I clinical trials. Cancer Res. 2016, 76, 1224. [Google Scholar] [CrossRef]
- Jeay, S.; Chène, P.; Ferretti, S.; Furet, P.; Gruenenfelder, B.; Guagnano, V.; Guerreiro, N.; Halilovic, E.; Hofmann, F.; Kallen, J. NVP-HDM201: Cellular and in vivo profile of a novel highly potent and selective PPI inhibitor of p53-Mdm2. Cancer Res. 2016, 76, 1225. [Google Scholar] [CrossRef]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the p53-MDM2 Inhibitor HDM201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef][Green Version]
- Vaupel, A.; Holzer, P.; Ferretti, S.; Guagnano, V.; Kallen, J.; Mah, R.; Masuya, K.; Ruetz, S.; Rynn, C.; Schlapbach, A.; et al. In vitro and in vivo characterization of a novel, highly potent p53-MDM2 inhibitor. Bioorg. Med. Chem. Lett. 2018, 28, 3404–3408. [Google Scholar] [CrossRef]
- Stein, E.; Chromik, J.; DeAngelo, D.J.; Chatterjee, M.; Noppeney, R.; Vos, F.d.; Minami, H.; Jeay, S.; Meille, C.; Halilovic, E. Abstract CT152: Phase I dose-and regimen-finding study of NVP-HDM201 in pts with advanced TP53 wt acute leukemias. Cancer Res. 2017, 77, CT152. [Google Scholar] [CrossRef]
- Hyman, D.M.; Chatterjee, M.; de Vos, F.; Lin, C.-C.; Suarez, C.; Tai, D.; Cassier, P.; Yamamoto, N.; de Weger, V.A.; Jeay, S. Optimizing the therapeutic index of HDM2 inhibition: Results from a dose-and regimen-finding phase I study of NVP-HDM201 in pts with TP53 wt advanced tumors. In Proceedings of the Cancer Research, Washington, DC, USA, 1–5 April 2017. [Google Scholar]
- Arnhold, V.; Schmelz, K.; Proba, J.; Winkler, A.; Wünschel, J.; Toedling, J.; Deubzer, H.E.; Künkele, A.; Eggert, A.; Schulte, J.H.; et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget 2018, 9, 2304–2319. [Google Scholar] [CrossRef] [PubMed]
- da Mota, V.H.S.; Freire de Melo, F.; de Brito, B.B.; da Silva, F.A.F.; Teixeira, K.N. Molecular docking of DS-3032B, a mouse double minute 2 enzyme antagonist with potential for oncology treatment development. World J. Clin. Oncol. 2022, 13, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, J.; Nakamaru, K.; Seki, T.; Tazaki, K.; Kojima, K.; Chachad, D.; Zhao, R.; Heese, L.; Ma, W.; Ma, M.C.J.; et al. Predictive Gene Signatures Determine Tumor Sensitivity to MDM2 Inhibition. Cancer Res. 2018, 78, 2721–2731. [Google Scholar] [CrossRef][Green Version]
- Ananthapadmanabhan, V.; Frost, T.C.; Soroko, K.M.; Knott, A.; Magliozzi, B.J.; Gokhale, P.C.; Tirunagaru, V.G.; Doebele, R.C.; DeCaprio, J.A. Milademetan is a highly potent MDM2 inhibitor in Merkel cell carcinoma. JCI Insight 2022, 7, e160513. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Fujiwara, Y.; Nakano, K.; Shimizu, T.; Tomomatsu, J.; Koyama, T.; Ogura, M.; Tachibana, M.; Kakurai, Y.; Yamashita, T.; et al. Safety and pharmacokinetics of milademetan, a MDM2 inhibitor, in Japanese patients with solid tumors: A phase I study. Cancer Sci. 2021, 112, 2361–2370. [Google Scholar] [CrossRef]
- Sekiguchi, N.; Kasahara, S.; Miyamoto, T.; Kiguchi, T.; Ohno, H.; Takagi, T.; Tachibana, M.; Sumi, H.; Kakurai, Y.; Yamashita, T.; et al. Phase I dose-escalation study of milademetan in patients with relapsed or refractory acute myeloid leukemia. Int. J. Hematol. 2023, 117, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Bauer, T.M.; Schwartz, G.K.; Weise, A.M.; LoRusso, P.; Kumar, P.; Tao, B.; Hong, Y.; Patel, P.; Lu, Y.; et al. A First-in-Human Phase I Study of Milademetan, an MDM2 Inhibitor, in Patients with Advanced Liposarcoma, Solid Tumors, or Lymphomas. J. Clin. Oncol. 2023, 41, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Golestanian, S.; Sharifi, A.; Popowicz, G.M.; Azizian, H.; Foroumadi, A.; Szwagierczak, A.; Holak, T.A.; Amanlou, M. Discovery of novel dual inhibitors against Mdm2 and Mdmx proteins by in silico approaches and binding assay. Life Sci. 2016, 145, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Lemos, A.; Leão, M.; Soares, J.; Palmeira, A.; Pinto, M.; Saraiva, L.; Sousa, M.E. Medicinal Chemistry Strategies to Disrupt the p53-MDM2/MDMX Interaction. Med. Res. Rev. 2016, 36, 789–844. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-type TP53. Clin. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef]
- Carvajal, L.A.; Neriah, D.B.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef][Green Version]
- Pairawan, S.; Zhao, M.; Yuca, E.; Annis, A.; Evans, K.; Sutton, D.; Carvajal, L.; Ren, J.G.; Santiago, S.; Guerlavais, V.; et al. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 2021, 23, 29. [Google Scholar] [CrossRef]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osório, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; Codon Publications: Brisbane, Australia, 2017; pp. 197–241. [Google Scholar]
- Nickoloff, J.A.; Taylor, L.; Sharma, N.; Kato, T.A. Exploiting DNA repair pathways for tumor sensitization, mitigation of resistance, and normal tissue protection in radiotherapy. Cancer Drug Resist. 2021, 4, 244–263. [Google Scholar] [CrossRef]
- Toulany, M. Targeting DNA Double-Strand Break Repair Pathways to Improve Radiotherapy Response. Genes 2019, 10, 25. [Google Scholar] [CrossRef][Green Version]
- Liu, Y.P.; Zheng, C.C.; Huang, Y.N.; He, M.L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef][Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- O’Brien, M.A.; Kirby, R. Apoptosis: A review of pro-apoptotic and anti-apoptotic pathways and dysregulation in disease. J. Vet. Emerg. Crit. Care 2008, 18, 572–585. [Google Scholar] [CrossRef]
- Bagnyukova, T.V.; Serebriiskii, I.G.; Zhou, Y.; Hopper-Borge, E.A.; Golemis, E.A.; Astsaturov, I. Chemotherapy and signaling. Cancer Biol. Ther. 2010, 10, 839–853. [Google Scholar] [CrossRef][Green Version]
- Sun, Y.; Liu, Y.; Ma, X.; Hu, H. The Influence of Cell Cycle Regulation on Chemotherapy. Int. J. Mol. Sci. 2021, 22, 6923. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.; Turchi, J.J. Chemotherapy induced DNA damage response. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Van Den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef]
- Swift, L.; Golsteyn, R. Genotoxic Anti-Cancer Agents and Their Relationship to DNA Damage, Mitosis, and Checkpoint Adaptation in Proliferating Cancer Cells. Int. J. Mol. Sci. 2014, 15, 3403–3431. [Google Scholar] [CrossRef][Green Version]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 Interaction for Cancer Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef][Green Version]
- Shen, H.; G Maki, C. Pharmacologic Activation of p53 by Small-Molecule MDM2 Antagonists. Curr. Pharm. Des. 2011, 17, 560–568. [Google Scholar] [CrossRef][Green Version]
- Nag, S.; Zhang, X.; Srivenugopal, K.S.; Wang, M.H.; Wang, W.; Zhang, R. Targeting MDM2-p53 Interaction for Cancer Therapy: Are We There Yet? Curr. Med. Chem. 2014, 21, 553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gao, H.; Ji, Y.; Zhou, Q.; Du, Z.; Tian, L.; Jiang, Y.; Yao, K.; Zhou, Z. Targeting p53–MDM2 interaction by small-molecule inhibitors: Learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol. 2022, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.I.; Jones, S.N. Regulation of the Mdm2-p53 signaling axis in the DNA damage response and tumorigenesis. Transl. Cancer Res. 2016, 5, 707–724. [Google Scholar] [CrossRef][Green Version]
- Klein, A.M.; De Queiroz, R.M.; Venkatesh, D.; Prives, C. The roles and regulation of MDM2 and MDMX: It is not just about p53. Genes Dev. 2021, 35, 575–601. [Google Scholar] [CrossRef]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef][Green Version]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feroz, W.; Sheikh, A.M.A. Exploring the multiple roles of guardian of the genome: P53. Egypt. J. Med. Hum. Genet. 2020, 21, 49. [Google Scholar] [CrossRef]
- Miles, X.; Vandevoorde, C.; Hunter, A.; Bolcaen, J. MDM2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front. Oncol. 2021, 11, 703442. [Google Scholar] [CrossRef] [PubMed]
- Welliver, M.X.; Tine, B.A.V.; Houghton, P.; Rudek, M.A.; Pollock, R.E.; Kane, J.M.; Schwartz, G.K.; Zhang, P.; Kirsch, D.G.; Wakely, P.; et al. MDM2 inhibitor AMG-232 and radiation therapy in treating patients with soft tissue sarcoma with wild-type TP53: A phase IB study (NRG-DT001). J. Clin. Oncol. 2019, 37, TPS11076. [Google Scholar] [CrossRef]
- Me, P. Mdm2 in the response to radiation. Mol. Cancer Res. MCR 2004, 2, 9–19. [Google Scholar]
- Geske, F.J.; Nelson, A.C.; Lieberman, R.; Strange, R.; Sun, T.; Gerschenson, L.E. DNA repair is activated in early stages of p53-induced apoptosis. Cell Death Differ. 2000, 7, 393–401. [Google Scholar] [CrossRef][Green Version]
- Koo, N.; Sharma, A.K.; Narayan, S. Therapeutics Targeting p53-MDM2 Interaction to Induce Cancer Cell Death. Int. J. Mol. Sci. 2022, 23, 5005. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, P.J.; Javaid, A.M.; Swick, A.D.; Werner, L.R.; Nickel, K.P.; Sampene, E.; Hu, R.; Ong, I.M.; Bruce, J.Y.; Hartig, G.K.; et al. Radiosensitization of Adenoid Cystic Carcinoma with MDM2 Inhibition. Clin. Cancer Res. 2017, 23, 6044–6053. [Google Scholar] [CrossRef][Green Version]
- Luo, H.; Yount, C.; Lang, H.; Yang, A.; Riemer, E.C.; Lyons, K.; Vanek, K.N.; Silvestri, G.A.; Schulte, B.A.; Wang, G.Y. Activation of p53 with Nutlin-3a radiosensitizes lung cancer cells via enhancing radiation-induced premature senescence. Lung Cancer 2013, 81, 167–173. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mladek, A.C.; Gupta, S.; Kim, M.; Mohammad, A.S.; Bakken, K.K.; He, H.; Hu, Z.; Burgenske, D.M.; Carlson, B.L.; Elmquist, W.F.; et al. Abstract C051: MDM2 inhibitor KRT-232 extends survival in glioblastoma patient-derived xenograft models. Mol. Cancer Ther. 2019, 18, C051. [Google Scholar] [CrossRef]
- Eischen, C.M. Role of Mdm2 and Mdmx in DNA repair. J. Mol. Cell Biol. 2023, 9, 69–73. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef][Green Version]
- Chen, L.; Agrawal, S.; Zhou, W.; Zhang, R.; Chen, J. Synergistic activation of p53 by inhibition of MDM2 expression and DNA damage. Proc. Natl. Acad. Sci. USA 1998, 95, 195–200. [Google Scholar] [CrossRef]
- Saiki, A.Y.; Caenepeel, S.; Yu, D.; Lofgren, J.A.; Osgood, T.; Robertson, R.; Canon, J.; Su, C.; Jones, A.; Zhao, X.; et al. MDM2 antagonists synergize broadly and robustly with compounds targeting fundamental oncogenic signaling pathways. Oncotarget 2014, 5, 2030–2043. [Google Scholar] [CrossRef][Green Version]
- Sarisozen, C.; Tan, Y.; Liu, J.; Bilir, C.; Shen, L.; Filipczak, N.; Porter, T.M.; Torchilin, V.P. MDM2 antagonist-loaded targeted micelles in combination with doxorubicin: Effective synergism against human glioblastoma via p53 re-activation. J. Drug Target. 2019, 27, 624–633. [Google Scholar] [CrossRef]
- Tong, H.; Zhao, K.; Zhang, J.; Zhu, J.; Xiao, J. YB-1 modulates the drug resistance of glioma cells by activation of MDM2/p53 pathway. Drug Des. Dev. Ther. 2019, 13, 317–326. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, X.; Tai, L.; Gao, J.; Qian, J.; Zhang, M.; Li, B.; Xie, C.; Lu, L.; Lu, W.; Lu, W. A stapled peptide antagonist of MDM2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation. J. Control. Release 2015, 218, 29–35. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Costa, B.; Bendinelli, S.; Gabelloni, P.; Da Pozzo, E.; Daniele, S.; Scatena, F.; Vanacore, R.; Campiglia, P.; Bertamino, A.; Gomez-Monterrey, I.; et al. Human Glioblastoma Multiforme: p53 Reactivation by a Novel MDM2 Inhibitor. PLoS ONE 2013, 8, e72281. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cai, S.; Bailey, B.J.; Reza Saadatzadeh, M.; Ding, J.; Tonsing-Carter, E.; Georgiadis, T.M.; Zachary Gunter, T.; Long, E.C.; Minto, R.E.; et al. Combination therapy in a xenograft model of glioblastoma: Enhancement of the antitumor activity of temozolomide by an MDM2 antagonist. J. Neurosurg. JNS 2017, 126, 446–459. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pi, L.; Rooprai, J.; Allan, D.S.; Atkins, H.; Bredeson, C.; Fulcher, A.J.; Ito, C.; Ramsay, T.; Stanford, W.L.; Sabloff, M.; et al. Evaluating dose-limiting toxicities of MDM2 inhibitors in patients with solid organ and hematologic malignancies: A systematic review of the literature. Leuk. Res. 2019, 86, 106222. [Google Scholar] [CrossRef]
- Sato, A.; Sunayama, J.; Matsuda, K.-I.; Seino, S.; Suzuki, K.; Watanabe, E.; Tachibana, K.; Tomiyama, A.; Kayama, T.; Kitanaka, C. MEK-ERK signaling dictates DNA-repair gene MGMT expression and temozolomide resistance of stem-like glioblastoma cells via the MDM2-p53 axis. Stem Cells 2011, 29, 1942–1951. [Google Scholar] [CrossRef]
- Felsberg, J.; Hentschel, B.; Kaulich, K.; Gramatzki, D.; Zacher, A.; Malzkorn, B.; Kamp, M.; Sabel, M.; Simon, M.; Westphal, M.; et al. Epidermal Growth Factor Receptor Variant III (EGFRvIII) Positivity in EGFR-Amplified Glioblastomas: Prognostic Role and Comparison between Primary and Recurrent Tumors. Clin. Cancer Res. 2017, 23, 6846–6855. [Google Scholar] [CrossRef][Green Version]
- Daniele, S.; Costa, B.; Zappelli, E.; Da Pozzo, E.; Sestito, S.; Nesi, G.; Campiglia, P.; Marinelli, L.; Novellino, E.; Rapposelli, S.; et al. Combined inhibition of AKT/mTOR and MDM2 enhances Glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Sci. Rep. 2015, 5, 9956. [Google Scholar] [CrossRef][Green Version]
- A Phase 1 Study Evaluating AMG 232 in Advanced Solid Tumors or Multiple Myeloma—Full Text View. 2023. Available online: https://ClinicalTrials.gov (accessed on 15 February 2023).
- 1Testing the Ability of AMG 232 (KRT 232) to Get into the Tumor in Patients with Brain Cancer—Full Text View. 2023. Available online: https://ClinicalTrials.gov (accessed on 15 February 2023).
- NCT Neuro Master Match-N²M² (NOA-20)—Full Text View. 2023. Available online: https://ClinicalTrials.gov (accessed on 15 February 2023).
- Phase 1 Study of the Dual MDM2/MDMX Inhibitor ALRN-6924 in Pediatric Cancer—Full Text View. 2023. Available online: https://ClinicalTrials.gov (accessed on 15 February 2023).
- A Study to Determine How BI 907828 is Taken up in the Tumor and to Determine the Highest Dose of BI 907828 that Could be Tolerated in Combination with Radiation Therapy in People with a Brain Tumor Called Glioblastoma—Full Text View. 2023. Available online: https://ClinicalTrials.gov (accessed on 15 February 2023).
- Schöffski, P.; Lahmar, M.; Lucarelli, A.; Maki, R.G. Brightline-1: Phase II/III trial of the MDM2-p53 antagonist BI 907828 versus doxorubicin in patients with advanced DDLPS. Future Oncol. 2023, 19, 621–629. [Google Scholar] [CrossRef]
- Montesinos, P.; Beckermann, B.M.; Catalani, O.; Esteve, J.; Gamel, K.; Konopleva, M.Y.; Martinelli, G.; Monnet, A.; Papayannidis, C.; Park, A.; et al. MIRROS: A randomized, placebo-controlled, Phase III trial of cytarabine ± idasanutlin in relapsed or refractory acute myeloid leukemia. Future Oncol. 2020, 16, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.Y.; Röllig, C.; Cavenagh, J.; Deeren, D.; Girshova, L.; Krauter, J.; Martinelli, G.; Montesinos, P.; Schäfer, J.A.; Ottmann, O.; et al. Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: Results of the MIRROS trial. Blood Adv. 2022, 6, 4147–4156. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| NCT Number | Title | MDM2 Inhibitor | Status | Condition | Ref. |
|---|---|---|---|---|---|
| NCT01723020 | A Phase 1 Study Evaluating AMG 232 in Advanced Solid Tumors or Multiple Myeloma | AMG 232 | Completed | Advanced Solid Tumors Glioblastoma Multiple Myeloma | [152] |
| NCT03107780 | Testing the Ability of AMG 232 (KRT 232) to Infiltrate the Tumor in Patients With Brain Cancer | Navtemadlin (KRT-232/AMG-232) | Suspended | Glioblastoma Gliosarcoma MGMT-Unmethylated Glioblastoma Recurrent Glioblastoma | [153] |
| NCT03158389 | NCT Neuro Master Match—N2M2 (NOA-20) (N2M2) | Idasanutlin (RG7388) | Recruiting | Glioblastoma, Adult | [154] |
| NCT03654716 | Phase 1 Study of the Dual MDM2/MDMX Inhibitor ALRN-6924 in Pediatric Cancer | ALRN-6924 | Recruiting | Leukemia Brain Tumor Solid Tumor Lymphoma | [155] |
| NCT05376800 | A Study to Determine How BI 907828 is Taken up in the Tumor and to Determine the Highest Dose of BI 907828 That Could be Tolerated in Combination With Radiation Therapy in People With a Brain Tumor Called Glioblastoma | BI 907828 | Recruiting | Glioblastoma | [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellot Ortiz, K.I.; Rechberger, J.S.; Nonnenbroich, L.F.; Daniels, D.J.; Sarkaria, J.N. MDM2 Inhibition in the Treatment of Glioblastoma: From Concept to Clinical Investigation. Biomedicines 2023, 11, 1879. https://doi.org/10.3390/biomedicines11071879
Pellot Ortiz KI, Rechberger JS, Nonnenbroich LF, Daniels DJ, Sarkaria JN. MDM2 Inhibition in the Treatment of Glioblastoma: From Concept to Clinical Investigation. Biomedicines. 2023; 11(7):1879. https://doi.org/10.3390/biomedicines11071879
Chicago/Turabian StylePellot Ortiz, Karolina I., Julian S. Rechberger, Leo F. Nonnenbroich, David J. Daniels, and Jann N. Sarkaria. 2023. "MDM2 Inhibition in the Treatment of Glioblastoma: From Concept to Clinical Investigation" Biomedicines 11, no. 7: 1879. https://doi.org/10.3390/biomedicines11071879