



by
, , , , , , and

Genes 2023, 14(9), 1749; https://doi.org/10.3390/genes14091749 (registering DOI) - 01 Sep 2023
Abstract
Habenaria dentata has medicinal and ornamental value, but the number of wild populations is decreasing dramatically. Thus, conducting research on its genetic diversity and structure is necessary to provide a basis for its conservation. This study aimed to explore the genetic diversity of
[...] Read more.
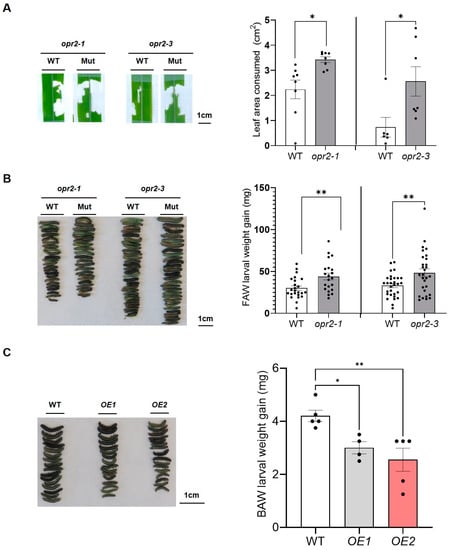
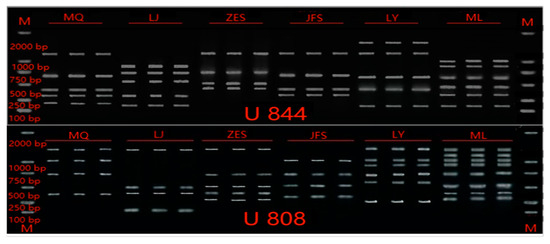
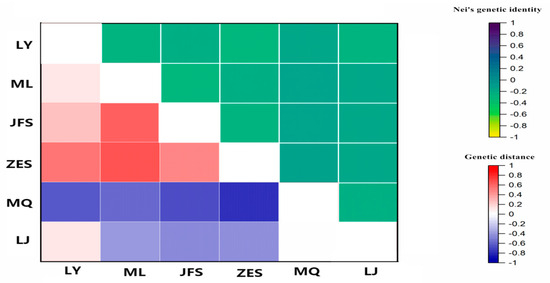
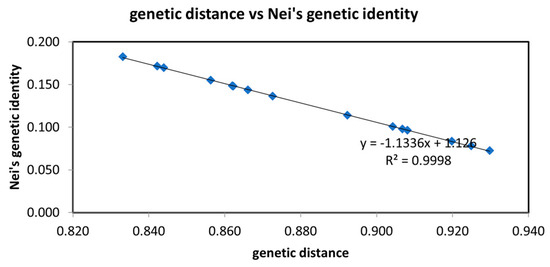
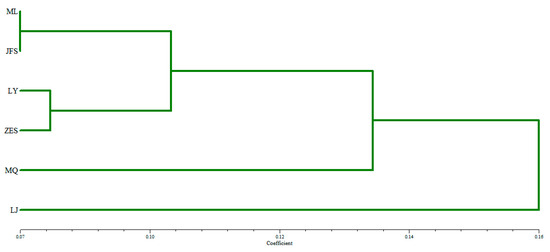
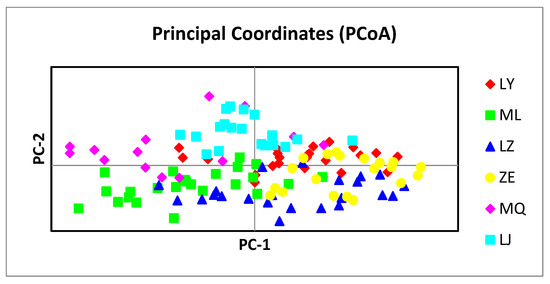
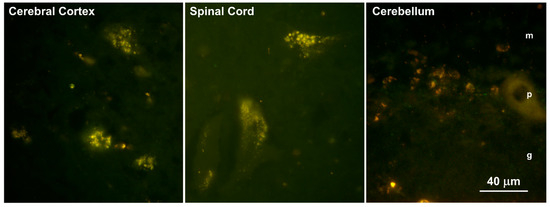
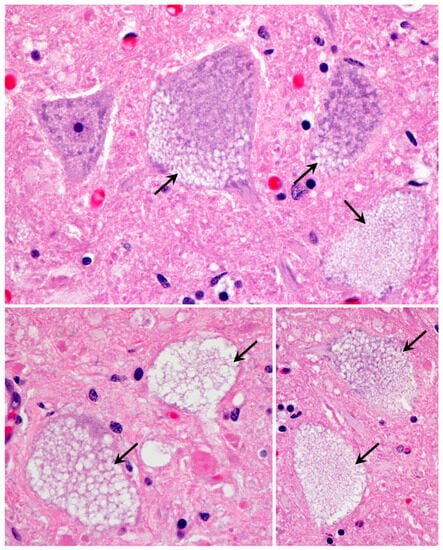
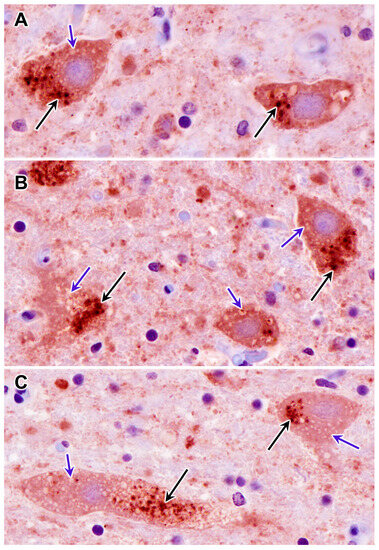
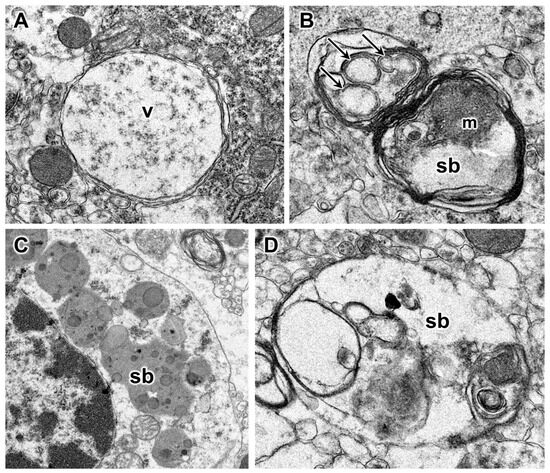
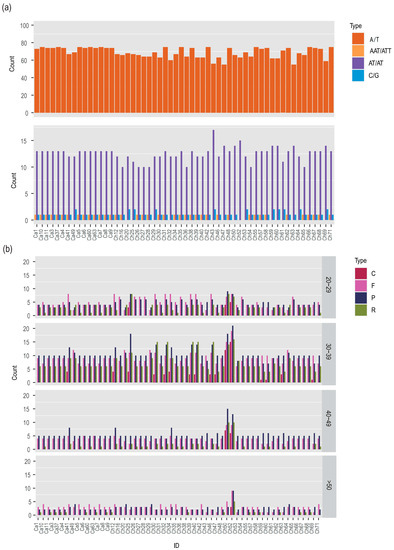
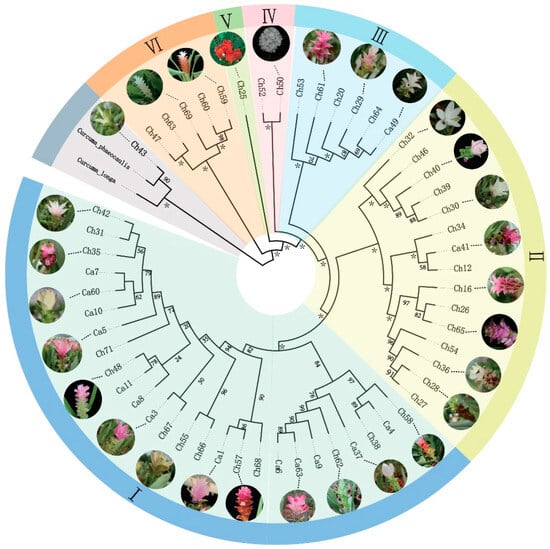
Habenaria dentata has medicinal and ornamental value, but the number of wild populations is decreasing dramatically. Thus, conducting research on its genetic diversity and structure is necessary to provide a basis for its conservation. This study aimed to explore the genetic diversity of the wild plant H. dentata and protect and optimize its wild resources. The genetic diversity of 133 samples from six wild populations of H. dentata was analyzed using Inter Simple Sequence Repeat molecular markers to provide a scientific basis for the screening of improved germplasm resources. The results showed that the average number of alleles was 1.765, the average number of effective alleles was 1.424, the average Nei’s gene diversity index was 0.252, the average Shannon diversity index was 0.381, and the average percentage of polymorphic loci was 76.499%. The variation within the populations was 77.34%, and the variation between the populations was 22.66%. The gene flow was 1.705, which was greater than 1. The results of the cluster analysis showed that the six populations were mainly divided into four clusters and were not classified according to their geographical location. There was no significant correlation between the geographical location and genetic distance between the populations (r = 0.557, p > 0.05). The genetic diversity of H. dentata is high. Among the six wild populations, the genetic diversity of the Mulun population was the highest and this population can be used as a key protection unit. The study on the genetic diversity of H. dentata can not only reveal the reasons for the decrease in the number of individuals in the population to a certain extent, and put forward the protection strategy, but also provide a scientific basis for the breeding of excellent seed resources.
Full article
(This article belongs to the Section Plant Genetics and Genomics)
►
Show Figures

Figure 1












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}